- Development of Cleaning Procedures by Research and Development groups (if it exists)
- Validation of Cleaning Procedures by Technical or Quality groups
- Application of Cleaning Processes by Operations/ Manufacturing organizations
A perceptive reader of this article may have noticed that those responsible for pivotal parts of the ECP continuum belong to different groups. Therefore an inevitable question, “Who is the Process Owner?” is typically and intuitively answered, “Operations or Manufacturing,” since they perform a majority of the continuum’s work. One might think, “They must be the owners since they clean equipment all the time.” Although, upon further examination it is clear that typically Operations or Manufacturing departments do not develop the process and do not qualify that it consistently delivers a quality result. Therefore, when cleaning fails it is typically assumed that it was not carried out correctly. Human error is often cited as a cause of deviations and nonconformances. Most of the time, despite extensive global regulations and a voluminous technical literature library on the subject, a failure of a Cleaning Process is not a human error but a failure to recognize the Cleaning as an official process and treat it as such. Instead of pursuit of process understanding, these failures cause organizations to perceive ECP as a “necessary evil"—something to tolerate mostly due to regulations. Many personnel may try to avoid it altogether and might argue that it is not a value added activity. However, it is obvious that this kind of philosophy is wrong and Cleaning must be recognized as an important and necessary part of pharmaceutical manufacturing. If one does not develop an appropriate method for Cleaning Process residuals, does not appropriately validate this method, and does not assure that it is still viable throughout the lifecycle of the product, one is adding another unknown variable into an actual manufacturing process. As stated by Deming, “uncontrolled variation is the enemy of quality.”
Therefore, the goal of this article is to help practitioners in development, utilization, and maintenance of Cleaning Validation programs so that they can reduce process variability thus answering the question—How Clean is Clean in Drug Manufacturing? To achieve this goal we will touch upon an important aspect of Cleaning Validation that gained traction in the last several years due to the implementation of risk-based lifecycle approach to Cleaning Validation built on principles of ICH Q8, 9, 10 Guidance, FDA Guidance for Industry: Process Validation, as well as EU Annex 15: Qualification and Validation (recent draft).
The subject we will discuss is development of limits for the process residue. Knowledge of the limits-setting strategies should help in gauging one’s Cleaning Validation program.
Establishing Limits
The subject of soil residue limits setting is of an utmost importance because it is a measure of the Cleaning Process effectiveness and consistency. We measure the success of a Cleaning Validation study by assuring that we meet predetermined criteria based on removal of the soil to a level below an established limit. Therefore establishing limits is one of the pivotal steps in the Cleaning Validation continuum. Although there are many sources on this topic and we can specifically mention a few excellent references for various methods of setting soil residual limits as well as history of the subject, our goal would be to employ science and knowledge into this exercise. First, we will briefly talk about Health-Based Exposure Limits since this subject has been debated for a few years and readers may benefit from some level of demystification. We will only summarize a few important points to consider when evaluating Health-Based Exposure Limits. There are several major terms being currently used globally for these limits. They are listed in Table 1 along with their respective sources and some notes that describe their intentions and use.Table 1. Health-Based Exposure Limits Terms
 Zoom In
Zoom InTable 2. NOEL vs. NO[A]EL

Cleaning Validation practitioners should consider these differences and utilize a toxicologist or a person with adequate training in pharmacology and toxicology to develop and document Health-Based Exposure Limits Assessments for soil residual limit calculations.
Although undisputedly important, the Health-Based Exposure Limits should not be the only ones utilized for measuring of a Cleaning Process. In order to gain a thorough understanding of the level of cleanliness needed, it is recommended to utilize the limits-setting practice using multiple levels. These levels are summarized in Table 3.
Table 3. Risk Levels for Setting Cleaning Validation Limits
 Zoom In
Zoom In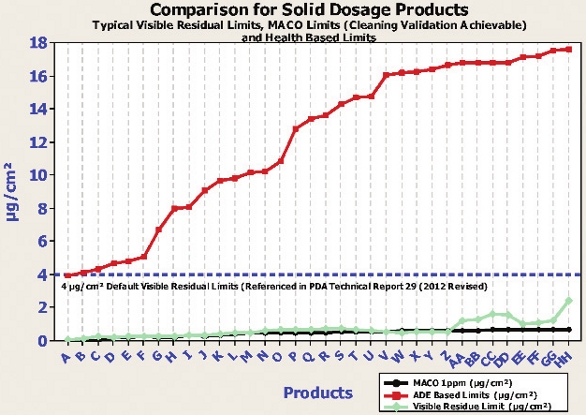 Zoom In
Zoom In
No comments:
Post a Comment